Displasia del tessuto osseo. Displasia fibrosa: cause, sintomi, metodi di trattamento
è una lesione ossea in cui un'area di tessuto osseo normale viene sostituita da tessuto connettivo comprendente trabecole ossee. Appartiene alla categoria delle malattie simil-tumorali, può essere locale o diffusa, interessando una o più ossa. Si manifesta con dolore, deformazione, accorciamento o allungamento del segmento e fratture patologiche. La diagnosi viene fatta sulla base della radiografia, della risonanza magnetica, della TC e di altri studi. Il trattamento è solitamente chirurgico: resezione dell'area interessata dell'osso con sostituzione del difetto.
ICD-10
M85.0 M85.4 M85.5

informazioni generali
La displasia fibrosa (malattia di Lichtenstein, malattia di Lichtenstein-Jaffe o malattia di Lichtenstein-Braizow) è una malattia scheletrica sistemica. I sintomi di solito iniziano durante l'infanzia, ma è possibile un'esordio più tardivo. La letteratura descrive casi in cui la displasia fibrosa monostotica è stata diagnosticata per la prima volta in persone in età pensionabile. Le donne si ammalano più spesso degli uomini. Possibile degenerazione in tumore benigno; la malignità è rara.
La malattia fu descritta per la prima volta nella prima metà del XX secolo. Nel 1927, il chirurgo russo Braitsov fece un rapporto sui segni clinici, microscopici e radiologici della degenerazione ossea fibrosa focale. Nel 1937, Albright descrisse la displasia fibrosa multifocale associata a disturbi endocrini e caratteristici cambiamenti della pelle. Nello stesso anno Albrecht descrisse una displasia multifocale associata a pubertà precoce e pigmentazione cutanea poco chiara. Poco dopo, Jaffe e Lichtenstein studiarono le lesioni focali singole e pubblicarono risultati sulle cause della loro insorgenza.
Cause
La displasia fibrosa è classificata come una malattia simil-tumorale, ma non è un vero tumore osseo. Si verifica a causa dello sviluppo improprio del mesenchima osteogenico (tessuto da cui si forma successivamente l'osso). Le ragioni dello sviluppo non sono chiare; non si può escludere una predisposizione genetica.
Classificazione
Tipicamente, con la forma poliostotica, si osserva un danno alle ossa tubolari: tibia, femore, perone, omero, radio e ulna. Delle ossa piatte, le ossa pelviche, le ossa del cranio, la colonna vertebrale, le costole e la scapola sono quelle più spesso colpite. Spesso viene rilevato un danno alle ossa delle mani e dei piedi, mentre le ossa del polso rimangono intatte. Il grado di deformazione dipende dalla posizione dei fuochi della displasia fibrosa. Quando si verifica un processo nelle ossa tubolari degli arti superiori, di solito si osserva solo la loro espansione a forma di mazza. Quando sono colpite le falangi, le dita si accorciano e sembrano “mozzate”.
Le ossa degli arti inferiori si piegano sotto il peso del corpo e si verificano deformazioni caratteristiche. Il femore è particolarmente deformato; nella metà dei casi si rileva il suo accorciamento. A causa della progressiva curvatura delle sezioni prossimali, l'osso assume la forma di un boomerang (bastone da pastore, mazza da hockey), il grande trocantere si “muove” verso l'alto, raggiungendo talvolta il livello delle ossa pelviche. Il collo del femore si deforma e si verifica zoppia. L'accorciamento della coscia può variare da 1 a 10 cm.
Quando si forma una lesione nel perone non si verifica alcuna deformazione dell'arto; se è interessata la tibia si può osservare una curvatura a sciabola della tibia o una crescita più lenta dell'osso in lunghezza. L'accorciamento è solitamente meno pronunciato rispetto a una lesione al femore. La displasia fibrosa dell'ileo e dell'ischio provoca la deformazione dell'anello pelvico. Ciò, a sua volta, influisce negativamente sulla colonna vertebrale, causando cattiva postura, scoliosi o cifosi. La situazione è aggravata se il processo colpisce contemporaneamente il femore e le ossa pelviche, poiché in questi casi l'asse del corpo viene ulteriormente interrotto e aumenta il carico sulla colonna vertebrale.
La forma monoossea procede più favorevolmente; non sono presenti manifestazioni patologiche extraossee. La gravità e la natura delle deformità variano notevolmente a seconda della localizzazione, delle dimensioni della lesione e delle caratteristiche della lesione (totale o intraossea). Dolore, zoppia e aumento dell'affaticamento possono verificarsi dopo aver caricato il segmento interessato. Come per la forma poliostotica, sono possibili fratture patologiche.
Diagnostica
La diagnosi viene fatta da un ortopedico-traumatologo sulla base del quadro clinico e dei dati radiografici. Nella fase iniziale, le pellicole radiografiche nell'area della diafisi o della metafisi dell'osso interessato rivelano zone che assomigliano all'aspetto al vetro smerigliato. Successivamente la zona interessata assume un caratteristico aspetto maculato: zone di compattamento si alternano a zone di schiarimento. La deformazione è chiaramente visibile. Quando viene rilevata una singola lesione, è necessario escludere lesioni ossee multiple, che nelle fasi iniziali possono essere asintomatiche, per cui i pazienti vengono indirizzati alla resezione segmentale dell'area ossea interessata all'interno del tessuto sano e alla sostituzione del difetto con una innesto osseo. Per una frattura patologica viene applicato un apparato di Ilizarov. In caso di lesioni multiple vengono adottate misure preventive per prevenire deformità e fratture patologiche.
Prognosi e prevenzione
La prognosi per la vita è favorevole. In assenza di trattamento, soprattutto nella forma poliostotica, possono verificarsi gravi deformità invalidanti. A volte i focolai di displasia degenerano in tumori benigni (tumore a cellule giganti o fibroma non ossificante). Negli adulti sono stati descritti diversi casi di trasformazione maligna in sarcoma osteogenico. Non esiste una prevenzione specifica a causa dell’eziologia poco chiara della malattia.
38199 0
Come sapete, la scienza e la storia non solo possono dimenticare, ma anche ricordare.
Nel 1927 V.R. Al 19 ° Congresso dei chirurghi russi, Braitsev fu il primo a fornire in dettaglio una descrizione accurata del quadro clinico, radiologico e microscopico delle ossa modificate e riferì sulla struttura microscopica del focolaio della displasia fibrosa. Credeva che la base della malattia fosse "la deviazione delle funzioni del mesenchima osteoblastico... Il mesenchima osteoblastico crea osso con una struttura incompleta". Pertanto, dobbiamo essere d'accordo con l'opinione di T.P. Vinogradova (1973), che ci sono molte più ragioni per assegnare il nome V.R. alla displasia ossea fibrosa. Braitsev, anziché chiamarla Lichtenstein o malattia di Lichtenstein-Jaffe, che ha solo chiarito e sviluppato ulteriormente le disposizioni della V.R. Braitseva.
VR Braitsev descrisse la displasia fibrosa anche nelle riviste “New Surgery” (1928) e “Archiv klinische Chirurgi” (1928), cioè 10 anni prima di J. Lichtenstein, che nel 1938 riferì di questa malattia e nel 1942 descrisse 15 delle sue osservazioni.
È necessario correggere questa ingiustizia storica: il merito di V.R. La scoperta da parte di Braitseva di una nuova entità nosologica – la displasia ossea fibrosa – è ovvia.
VR Braitsev nel 1927 al XIX Congresso dei chirurghi russi fece anche un rapporto sull'osteodistrofia locale - osteodistrofia fibrosa localisata (cistica). Ha detto: "I chirurghi sono particolarmente interessati al lato pratico della questione, ma l'opportunità delle misure chirurgiche per l'osteodistrofia locale può basarsi solo sulla completa chiarezza dell'essenza della malattia". Conoscendo bene la letteratura mondiale su questo tema, sulla base di tre proprie osservazioni, propone una nuova teoria originale sull'origine e l'essenza della malattia, la identifica come un'unità nosologica indipendente, dimostra le ragioni dello sviluppo del tessuto fibroso cisti e raccomanda metodi di trattamento. La descrizione del quadro morfologico della displasia fibrosa è insolitamente accurata. In conclusione, trae le seguenti conclusioni.
1. L'essenza dell'osteodistrofia fibrosa è la deviazione funzionale del mesenchima osteoblastico durante lo sviluppo osseo nel periodo embrionale, a seguito della quale, fin dall'inizio, si crea un osso peculiare con midollo osseo fibroso, capace di crescere e produrre “osteoide tessuti e ossa di tipo incompleto”.
2. Tale deviazione nella funzione del mesenchima osteoblastico può verificarsi in aree isolate di un singolo osso o può diffondersi all'intero osso e persino a molte ossa dello scheletro.
3. La crescita del tessuto fibroso è attiva, ma l'energia di crescita è diversa nei diversi casi. In alcuni casi, il processo procede silenziosamente e lentamente, in altri - rapidamente, accompagnato da un ampio polimorfismo cellulare, che lo avvicina morfologicamente al sarcomatoso.
4. Le cisti ossee solitarie, secondo i dati ottenuti da molti autori, si sviluppano a causa dell'osteodistrofia fibrosa dovuta all'edema e alla liquefazione delle escrescenze fibrose centrali, e anche, possibilmente, a causa di emorragie nel tessuto fibroso.
V. R. Braitsev consiglia di eseguire una resezione sottoperiostale longitudinale con sostituzione del difetto con autoinnesti, perché "il tessuto fibroso patologico, come si può vedere dall'esame istologico, penetra nel guscio osseo fino al periostio".
Chirurghi eminenti come I.I. Grekov, S.P. Fedorov, N.N. Petrov, ma dai loro discorsi è chiaro che hanno sottovalutato i dati unici ottenuti da V.R. Braitsev: la sua scoperta di una nuova unità nosologica. Tutti questi chirurghi, come N.N. Terebinsky e T.N. Krasnobaev, parlavano solo delle cisti ossee, che spesso incontravano e per le quali a volte eseguivano operazioni.
Sotto la nostra osservazione in ospedale c'erano 245 pazienti con displasia fibrosa; il numero di pazienti con lesioni poliostotiche era significativamente maggiore del numero di pazienti con processo monostotico che necessitavano di trattamento chirurgico.
Secondo la letteratura, le forme monoostotiche e poliostotiche di displasia fibrosa si osservano quasi altrettanto spesso, tuttavia, secondo M.K. Klimova (1970), la forma poliostotica è ancora un po' più comune, e anche M.V. Volkov (1968, 1985).
Clinica. I pazienti nascono raramente con gravi deformità scheletriche. I sintomi della displasia fibrosa compaiono solitamente durante l'infanzia e sono caratterizzati da diversità: si tratta di un dolore lieve, solitamente ai fianchi, o della comparsa di deformità e del suo aumento, o di una frattura patologica dovuta a un trauma grave e inadeguato, e la diagnosi corretta è non sempre realizzato.
Nella forma poliostotica della displasia fibrosa, sono più spesso colpiti la tibia, il femore, il perone, l'omero, il radio e l'ulna. La frequenza del danno (in ordine decrescente) delle ossa piatte: ossa pelviche, ossa del cranio, vertebre, costole, scapola. Le ossa del piede e della mano (ma non le ossa del polso) sono relativamente spesso colpite.
I bambini affetti dalla sindrome di Albright talvolta nascono con gravi deformità e, ovviamente, con una tipica macchia pigmentata. Dopo la prima manifestazione della malattia, sia i segni clinici che quelli radiologici possono progredire e la forma intraossea della malattia può poi svilupparsi in una forma che colpisce l'intero strato corticale o nell'area di uno dei focolai, più spesso nella parte superiore fine del femore o lungo tutta la diafisi, che indica varie attività del processo displastico. Le epifisi delle ossa, di regola, non sono interessate. La progressione del processo nei bambini e nei giovani è spesso accompagnata da fratture. Secondo le osservazioni di A.I. Snetkova (1984), in un paziente operato a 4 anni (resezione marginale, rimozione di tessuto fibroso, alloplastica ossea), dopo 7 anni si è osservata una ristrutturazione degli alloinnesti con zone di loro lisi, causata dalla crescita di nuovi focolai patologici. Pertanto, esiste uno schema programmato nello sviluppo della displasia: in alcuni pazienti si sviluppa tessuto fibroso displastico in aree dell'osso che precedentemente apparivano normali radiograficamente.
L.N. Furtseva et al. (1991) hanno rivelato disturbi significativi nella funzione glucocorticoide della corteccia surrenale in pazienti con displasia fibrosa: “Il livello di calcio in tutti i tipi di malattia è aumentato, ma non in proporzione all'entità del danno al tessuto osseo; allo stesso tempo, l'escrezione di calcio nelle urine è ridotta rispetto alla norma. La diminuzione è più pronunciata nella forma poliostotica che in quella monoostotica. Nelle forme limitate della malattia, la fosfaturia è ridotta nelle lesioni estese del tessuto osseo, si osserva solo una tendenza al ribasso; L'azoto amminico totale e l'idrossiprolina totale nelle urine aumentano nei processi estesi, e nella sindrome di Albright e nella forma poliostotica con lesioni diffuse, l'escrezione di aminoacidi è significativamente più elevata.
Sistema cardiovascolare: più spesso nei pazienti con forma poliostotica si osserva tachicardia sinusale - 96-140 al minuto, meno spesso si nota aritmia sinusale sull'ECG e nella maggior parte dei pazienti l'ipotensione arteriosa è 115/60 e persino 95/50 mm Hg, con In alcuni pazienti, il metabolismo nel muscolo cardiaco viene interrotto. Si nota un aumento della VES: nei pazienti con forma monoostotica - fino a 15-27 mm/h, con forma poliostotica - fino a 22-45 mm/h. Durante lo studio della funzione surrenale, è stato determinato il contenuto di 11-idrossicorticosteroidi (11-OX) nel plasma; Nei pazienti con forma poliostotica è stato rivelato un disturbo dello stato funzionale della corteccia surrenale, come evidenziato da un aumento meno pronunciato del livello di 11-OX totale e attivo nel plasma sanguigno e da una risposta indebolita o paradossale alla somministrazione dell’ormone adrenocorticotropo (ACTH).
Un paziente di 30 anni affetto dalla forma poliostotica morì durante un intervento chirurgico nel 1974 a causa di un improvviso calo della pressione sanguigna; il secondo, all'età di 19 anni, morì dopo un intervento chirurgico nel 1978 per insufficienza cardiaca polmonare progressiva. Durante l'esame patologico, oltre ai tipici cambiamenti nelle ossa, sono stati rilevati cambiamenti simili: ovaie policistiche, adenomatosi della corteccia surrenale, ghiandola tiroidea, ghiandola pituitaria anteriore e iperplasia del timo.
A.I. Morozov, V.P. Ivannikov (1972), studiando un caso di “malattia di Albright” in un paziente di 16 anni, identificò cambiamenti nelle strutture cerebrali profonde della linea mediana sull’elettroencefalogramma. A.G. Povarinsky, Z.K. Bystrov, con l'aiuto di studi encefalografici condotti parallelamente a studi neurologici, ha stabilito disfunzioni profonde e caratteristiche del cervello e l'instabilità dell'omeostasi cerebrale come risultato di un disturbo dei meccanismi regolatori situati nell'area delle strutture profonde. Tutto ciò ci ha costretto negli ultimi 20 anni ad esaminare attentamente i pazienti con displasia fibrosa, soprattutto con la forma poliostotica.
In un certo numero di pazienti, questa displasia si manifesta in modo latente. Abbiamo consultato un professore-chirurgo di 62 anni, nel quale per la prima volta è stata scoperta casualmente sulle radiografie la displasia fibrosa dell'intero femore sinistro e della tibia, e non abbiamo stabilito alcuna indicazione per il trattamento chirurgico. Non crediamo che tutti i pazienti affetti da displasia fibrosa debbano essere operati, soprattutto quelli con forma intraossea. Le indicazioni per l'intervento chirurgico sono il dolore causato da deformazione progressiva, fratture da stress, presenza di cisti con un forte assottigliamento dello strato corticale, zoppia, accorciamento dell'arto, compressione del midollo spinale, ecc. Se c'è una frattura nella sede della cisti, eseguiamo un trattamento conservativo, aspettiamo 8 mesi dopo la guarigione e se la cisti rimane della stessa dimensione o progredisce, operiamo.
M.V. Volkov (1985) distingue tra forme poliostotiche, monoossee e regionali di displasia fibrosa e in base alla natura dei cambiamenti nell'osso - focali e diffusi. Offriamo una classificazione clinica con una descrizione più dettagliata delle caratteristiche di ciascuna forma, utilizzando il concetto da noi proposto di "memoria della forma ossea", tuttavia, va tenuto presente che la displasia fibrosa è un processo patologico che presenta innumerevoli varianti e forme transitorie .
Classificazione clinica della displasia ossea fibrosa (secondo S.T. Zatsepin)
La classificazione proposta include le seguenti forme. I. Forma intraossea di displasia fibrosa: i focolai di tessuto fibroso possono essere singoli, multipli, occupare qualsiasi parte dell'osso o l'intero osso, tuttavia, lo strato corticale può essere assottigliato, ma mantiene una struttura normale - la forma delle ossa rimane corretto, poiché non vi è alcuna compromissione della memoria nelle forme ossee. Possono essere colpiti uno o più ossa di diversi segmenti dell'arto, ad es. il processo è monoostotico o poliostotico.
Un intervento adeguato è la rimozione completa del tessuto fibroso mediante resezione marginale dell'osso della lunghezza richiesta, rimozione del tessuto fibroso e sostituzione della cavità con omoinnesti ossei conservati. L'intervento può essere considerato radicale se le pareti della cavità sono intatte il tessuto fibroso individuato viene accuratamente lavorato con uno scalpello. In questa forma, le cisti si sviluppano spesso al centro di masse fibrose; le cisti raggiungono la corticale assottigliandola, dando spesso luogo a fratture patologiche (Fig. 15.1). Più spesso ricorrono a tattiche in due fasi: 1) viene applicata la trazione scheletrica - i frammenti guariscono bene, poiché lo strato corticale e il periostio sono normali; spesso le cavità cistiche scompaiono e, in caso contrario, 2) eseguire la resezione marginale, la rimozione delle masse fibrose, l'alloplastica ossea o eseguire un'operazione secondo la nostra tecnica (vedi sotto).
II. Il processo patologico coinvolge tutti gli elementi dell'osso: l'area del canale midollare, lo strato corticale, la spongiosi delle metafisi, le ossa lunghe sono spesso colpite per tutta la lunghezza, ma la gravità del processo patologico varia; di solito è una lesione poliostotica. Il danneggiamento di tutti gli elementi che compongono l'osso (danno totale) riduce la sua resistenza meccanica, portando a deformazioni graduali e fratture per fatica. Con questa forma di displasia fibrosa SI ESPRESSA LA SINDROME DELLA MEMORIA OSSEA. Non esistono interventi chirurgici radicali adeguati (ad eccezione della rimozione dell’intero osso interessato – vedere paragrafo 15.2). Molto utilizzate sono le osteotomie correttive ortopediche con alloplastica e strutture metalliche.
Riso. 15.1. Displasia fibrosa. Forma intraossea. a — parte della lesione è occupata da una cisti; b — operazione secondo S.T Zatsepin per una frattura ripetuta: le estremità dei frammenti sono esposte; rimuovere il tessuto fibroso con uno scalpello, cucchiai e alesatori; attraverso l'apertura del canale del midollo osseo nel sito dell'osteotomia - una frattura; i frammenti vengono fissati e la cavità viene riempita con perone allogenico, introdotto prima prossimalmente, poi distalmente.
Crescite simili a tumori di focolai di tessuto fibroso possono raggiungere grandi dimensioni, il che indica una maggiore massa del processo, ma sono raramente osservate.
III. Forme tumorali di displasia fibrosa.
IV. La sindrome di Albright è una forma speciale di displasia, quando, insieme alla forma poliostotica o quasi generalizzata - displasia ossea fibrosa totale - si osservano numerosi disturbi endocrini con pubertà precoce nelle donne, campi di pigmentazione sulla pelle, proporzioni corporee alterate e spesso bassa statura; gravi deformazioni delle ossa degli arti, del bacino, della colonna vertebrale, delle ossa della base del cranio, cambiamenti pronunciati nel sistema cardiovascolare e in altri sistemi corporei. Durante la vita, il processo progredisce, le deformazioni ossee aumentano gradualmente. La sindrome della memoria della forma ossea compromessa è pronunciata.
Esistono molte varianti di questa forma della malattia; nessun paziente ne ripete un'altra, ma è necessariamente diversa in qualche modo. Lo studio della struttura morfologica del tessuto fibroso e osseo patologico in pazienti con questa forma di displasia ha mostrato che le varie manifestazioni cliniche di questa displasia corrispondono a un'ampia varietà di modelli istologici, quindi il compito dei ricercatori è quello di studiare e confrontare clinico-radiomorfologico , che consentirà senza dubbio di identificare sottogruppi di pazienti.
V. La displasia ossea fibroso-cartilaginea come forma speciale nel nostro paese è stata isolata e descritta da M.A. Berglezov e N.G. Shulyakovskaya nel 1963, che osservò un paziente con un quadro morfologico clinico e radiografico pronunciato. Nelle osservazioni descritte, le manifestazioni di displasia cartilaginea vengono alla ribalta, i casi di sviluppo del condrosarcoma non sono rari;
VI. Il fibroma calcificante delle ossa lunghe appartiene a un tipo speciale di displasia fibrosa, descritta nel 1958 da N.E. Schlitter, R.L. Kempsom (1966), che lo studiò al microscopio elettronico.
QUELLO. Goerghen et al. (1977) descrissero 2 pazienti, uno dei quali ebbe una recidiva dopo il curettage e fu resecato insieme al periostio per 10 cm, ma dopo 1 anno fu notata una piccola recidiva all'estremità della parte inferiore della tibia - questo è stata l'unica osservazione in caso di recidiva del tumore. Sono stati descritti un totale di 8 tumori di questo tipo, localizzati nella tibia e un tumore nell'omero. La struttura istologica dei tumori è identica a quella osservata nelle ossa del cranio facciale e delle mascelle.
Trattamento. Nei pazienti con displasia fibrosa, il trattamento conservativo non viene utilizzato da nessuno degli autori a noi noti. Siamo stati costretti a utilizzare questo trattamento in 8 pazienti affetti da displasia fibrosa, nei quali le ossa degli arti, del bacino, della colonna vertebrale e della maggior parte delle costole erano alterate. Soffrivano forti dolori non solo quando stavano in piedi, ma anche quando respiravano. Per i pazienti in cui la 1a, 2a, 3a costola è alterata, potremmo alleviare il dolore mediante resezione della o delle costole più alterate (durante l'intervento è importante ricordare che, a causa della perdita di forza, affondano e si trovano più in profondità rispetto a quelle adiacenti normali). costolette). Quando sono colpite la maggior parte delle costole e delle vertebre, ciò non è possibile e le trattiamo con cicli ripetuti di iniezioni di calcitonina. In tutti i pazienti il dolore è diminuito, si è notato un miglioramento, ma per diversi anni non è stato possibile effettuare un trattamento sistematico su un gran numero di pazienti a causa della piccola quantità del farmaco. Poiché i pazienti con la forma poliostotica della displasia fibrosa presentano diversi cambiamenti nel sistema endocrino, siamo fiduciosi che sia necessario sviluppare una giustificazione scientifica per il trattamento conservativo. Poiché in un certo numero di pazienti con l'età, alcune lesioni, spesso dopo una frattura a questo livello, subiscono calcificazione e ossificazione, i metaboliti attivi della vitamina D3 e dei complessi dovrebbero essere inclusi nel complesso del trattamento conservativo.
Trattamento chirurgico. VR Braitsev (1927) usò sia la resezione marginale con sostituzione del difetto, sia la resezione totale - sottoperiostale con sostituzione del difetto con autoinnesti, poiché, secondo i suoi dati, il tessuto patologico penetra nello strato corticale fino al periostio. Negli anni successivi alla seconda “scoperta” di questa displasia, gli ortopedici iniziarono ad operare in modo meno radicale. Un'operazione tipica era la resezione marginale dello strato corticale dell'osso interessato durante l'intera lesione, ad es. spesso durante l'intera diafisi e metafisi, rimozione attenta con un cucchiaio affilato, uno scalpello semicircolare di tutte le masse fibrose e sostituzione del difetto con autologhi e negli ultimi 35 anni con allotrapianti ossei. 
Riso. 15.2. Deformazione del femore a “bastone da pastore”.
a — displasia fibrosa, deformazione della metà superiore del femore; b — Tecnica di S.T. Zatsepin: osteotomia correttiva all'apice della curvatura, la cavità viene riempita con perone allogenico e innesti corticali.
Nel 1978, correggendo una tipica deformità in varo del femore superiore a “bastone da pastore” (Fig. 15.2), abbiamo iniziato:
1) spostare medialmente l'estremità superiore del frammento inferiore, portandolo sotto il collo del femore, cioè accorciare la leva per correggere la posizione del frammento superiore, cioè teste, colli del grande trocantere;
2) sezionare ampiamente la capsula dell'articolazione dell'anca lungo la superficie superiore (non abbiamo mai dovuto tagliare i tendini dei muscoli glutei dal grande trocantere, come scrive A.I. Snetkov (1984), sebbene abbiamo operato pazienti adulti, e lui ha operato sui bambini e sugli adolescenti;
3) incrociare i tendini sottocutanei dei muscoli adduttori della coscia all'altezza dell'osso pubico;
4) rimuovere le masse fibrose dai frammenti superiori ed inferiori;
5) introdurre il perone allogenico nel canale distale e nel canale prossimale sotto forma di frammenti come fissatore endomidollare e materiale plastico pregiato;
6) fissare inoltre i frammenti con una piastra Trotsenko-Nuzhdin.
Si segnala in particolare quanto segue. In primo luogo, quando si eseguono osteotomie correttive in pazienti con displasia fibrosa, di regola, è meglio eseguire una semplice osteotomia trasversale piuttosto che una figurata, poiché lo strato corticale assottigliato si rompe rispetto al confronto e le spine risultanti formano una connessione ideale. In secondo luogo, il 13 novembre 1978, nella maggior parte dei pazienti, abbiamo abbandonato le resezioni marginali per rimuovere le masse fibrose dall’osso. Dopo aver eseguito un'osteotomia per correggere la deformità, senza eseguire la resezione marginale, distruggiamo il tessuto fibroso all'interno dell'osso con frese per espandere il canale quando si introducono chiodi intramidollari (come è noto, il loro diametro varia da 6 a 16 mm), e poi rimuovere le masse fibrose con curette ginecologiche di lunghezza sufficiente. Senza disturbare le pareti dei frammenti, abbiamo l'opportunità di inserire il perone intramidollare, talvolta insieme a un chiodo metallico, ottenere una fissazione affidabile e sostituire la cavità dopo aver rimosso il tessuto fibroso con osso conservato (Fig. 15.3). Riteniamo che se il tessuto fibroso viene completamente rimosso dall'osso, non è necessario il distacco del periostio per interrompere l'afflusso di sangue all'osso interessato, raccomandato da A.P. Berezhny, M.V Volkov, A.N.
M.V. Volkov, A.N. Dal 1982, Snetkov iniziò ad utilizzare massicce placche metalliche angolari sull'osso in pazienti con lesioni diffuse del femore lungo l'intera lunghezza, compreso il collo e la regione trocanterica del segmento, e sul lato opposto al fissatore, fu effettuato un massiccio allotrapianto corticale posizionato, cioè applicarono la tecnica e la fissazione ossea (con una leggera modifica) da noi proposta e iniziarono ad utilizzare con successo nel 1972 in pazienti con osteogenesi imperfetta. Come hanno dimostrato molti anni di osservazioni, il nostro metodo dei fissatori metallici esterni e dell’innesto osseo, che ha permesso di ottenere ottimi risultati nell’osteogenesi imperfetta, ha dato un effetto temporaneo in un gran numero di pazienti con forme diffuse di displasia fibrosa, e poi la il femore ha cominciato a piegarsi, le viti che fissavano l'innesto osseo si sono rotte o fuoriuscite dall'osso (abbiamo avuto l'opportunità di vedere questi pazienti che sono stati operati nel reparto pediatrico e poi sono stati ricoverati sotto la nostra supervisione nella clinica per adulti dell'ospedale CITO).
Vediamo la spiegazione di ciò nel fatto che nell'osteogenesi imperfetta viene conservata la memoria della forma dell'osso, mentre nelle lesioni diffuse delle ossa dovute a displasia fibrosa, le ossa perdono la memoria della forma e le deformazioni, di regola, si ripresentano. una laurea o l'altra.
Riteniamo quindi che definire radicali le operazioni attualmente in corso, come fanno alcuni autori, sia un errore. Bisogna ammettere che attualmente non è stato trovato alcun trattamento radicale per la displasia fibrosa. A volte trattare questi pazienti è molto difficile.
Ecco un esempio.
Paziente T., 27 anni. A causa delle gravi deformità delle braccia e delle gambe, i genitori abbandonarono la figlia e sua nonna la portò via dall'ospedale di maternità. A causa dei gravi cambiamenti causati dalla displasia fibrosa poliostotica, il paziente è stato costretto a letto per molti anni (Fig. 15.4). È stata ricoverata al CITO, dove abbiamo eseguito in sequenza 5 interventi chirurgici su entrambi i femori, la tibia destra, l'omero sinistro ed entrambe le ossa dell'avambraccio sinistro. Caratteristiche da sottolineare sono state l'uso di osteotomie segmentali, fissazione endomidollare con chiodo CITO e chirurgia plastica con adloinnesti corticali molto massicci (2/3 del diametro del femore), che svolgevano il ruolo di una stecca di fissazione biologica, con la quale frammenti del femore erano fusi per tutta la sua lunghezza. Questo è molto importante perché gli allotrapianti così massicci non si rimodellano mai completamente e quindi non si curvano; La connessione ossea su una superficie così ampia è più resistente delle viti metalliche. 
Riso. 15.3. Deformazione tagliente come un “bastone da pastore” 
Riso. 15.4. Displasia fibrosa poliostotica.
a, 6 gravi alterazioni e deformazioni di entrambi i femori e delle ossa della gamba sinistra e un tentativo di correggere la deformità (perno di Bogdanov, righello) d, e - dopo osteotomie correttive dei femori, fissazione con perni CITO e alloinnesti corticali massicci; e - ortesi esterne; il paziente 23 anni dopo l’intervento.
18/01/81 - tripla osteotomia correttiva del femore destro con fissazione endomidollare con perno, chirurgia plastica con allotrapianto corticale massiccio di lunghezza pari alla diafisi dell'osso del paziente. 28/03/81 - doppia osteotomia del femore sinistro, fissazione endomidollare con perno e alloplastica simile. 21/06/81 - osteotomia correttiva delle ossa della gamba sinistra nel terzo superiore, rimozione di masse fibrose, fissazione endomidollare, alloplastica. Esame morfologico: displasia fibrosa con ampio strato osteoide. 27/02/85 - osteotomia dell'omero sinistro a due livelli, asportazione di tessuto patologico, osteosintesi extra ed intramidollare con alloinnesti. 31.10.85 - osteotomia dell'ulna sinistra, asportazione di tessuto fibroso, alloplastica autologa. Le operazioni sono state eseguite da S.T. Zatsepin.
Il paziente è dotato di apparecchi ortopedici e lavora come corrispondente per un giornale locale.
Va ricordato che in alcuni pazienti con sindrome di Albright, i processi displastici nelle ossa progrediscono con l'età, la deformazione della colonna vertebrale, del cranio, del torace e delle ossa degli arti aumenta e le deformità corrette dalla chirurgia si ripresentano. Se durante l'intervento chirurgico durante la rimozione delle masse fibrose si osserva un aumento del sanguinamento, ciò significa che tali pazienti presentano gravi emorragie sia durante che dopo l'intervento. Un paziente, da noi operato 4 volte, nel periodo postoperatorio ha avuto tre forti emorragie, per le quali il chirurgo di turno ha dovuto operare e suturare le aree sanguinanti.
Abbiamo osservato la malignità della displasia fibrosa in 7 pazienti; Diamo un esempio. 
Riso. 15.5. Malignità della forma poliostotica della displasia fibrosa. La prima frattura del femore destro si è verificata all'età di 6 anni. La resezione marginale e la chirurgia plastica con allotrapianti corticali sono state eseguite due volte, seguite dall'allungamento del femore secondo Ilizarov. Si è sviluppato un sarcoma telangiectasico. Amputazione interiliaco-addominale.
Fedkushov Yu.I., 21 anni. 03.06.58 - resezione parziale longitudinale del terzo superiore e medio della coscia destra con omoplastica. Dal gennaio 1959 poteva camminare con un bastone. 23/03/60 - resezione longitudinale dello strato corticale lungo la superficie esterna nel terzo medio e inferiore della coscia destra, omoplastica. Nel 1967 - allungamento della coscia. Fatto da G.A. Ilizarov. Nel 1983 è comparso il dolore e poi il volume è aumentato di 4 cm nel terzo superiore e medio del femore destro. Biopsia: sarcoma telangiectasico (Fig. 15.5). Il 5 dicembre 1984 venne eseguita l'amputazione interiliaco-addominale. Vivo da 19 anni.
S.T.Zatsepin
Patologia ossea dell'adulto
La displasia fibrosa è una patologia che porta alla sostituzione dell'osso con tessuto connettivo, intervallato da trabecole ossee. Questo tipo di displasia ossea è classificata come una malattia tumorale. Esistono malattie monoossee (un osso è soggetto a deformazione) e molteplici tipi di malattie (la curvatura colpisce più ossa).
Le cause della malattia non sono completamente comprese. Molti medici suggeriscono una predisposizione ereditaria o un disturbo dello sviluppo intrauterino del feto. Consideriamo quali sintomi sono caratteristici della displasia ossea fibrosa e le regole per il trattamento della malattia.
Displasia fibrosa: descrizione della malattia
La displasia ossea fibrosa è anche chiamata malattia di Braitsev Liechtenstein in onore degli scopritori della malattia. Il chirurgo russo Braitsev ha descritto i segni della patologia nel suo rapporto e Lichtenstein ha esaminato i focolai delle lesioni poliostotiche e ha tratto conclusioni sulle cause della loro insorgenza. M85.0 – codice per displasia ossea fibrosa secondo ICD 10 (Classificazione internazionale delle malattie, 10a revisione).
L'osteodisplasia fibrosa è una deformazione sistemica dello scheletro, classificata come malattia tumorale, sebbene non sia una vera neoplasia. Si sviluppa a causa di una violazione della formazione del tessuto osteogenico - cellule da cui successivamente si formano le ossa della parte inferiore della gamba e della coscia.
Nella maggior parte dei casi, la displasia fibrosa degli arti inferiori si verifica durante l'infanzia e lo sviluppo successivo è meno comune; Le donne soffrono della patologia più spesso degli uomini.
Interessante!
Tipicamente, la displasia fibrosa è una neoplasia benigna (degenerazione in un tumore maligno) viene registrata molto raramente;
La patologia non deve essere confusa con la fibrodisplasia ossificante, una malattia rara che rappresenta la degenerazione irreversibile del tessuto scheletrico molle in osso. Le patologie colpiscono tendini, muscoli e fascia. La fibrodisplasia è caratterizzata da attacchi di gonfiore dei tessuti molli, che indicano il processo di degenerazione.
Tipi di displasia
Gli esperti classificano la displasia fibrosa in:
- Forma monossale: è interessata un'articolazione. Si verifica sia nei bambini che negli adulti;
- Poliostotico: la patologia colpisce diverse articolazioni su un lato del corpo. Diagnosticato durante l'infanzia e l'adolescenza, si sviluppa sullo sfondo di disturbi endocrini.
Un'altra classificazione popolare della displasia è stata proposta dal chirurgo clinico russo Zatsepin. Egli divide la patologia fibrotica in:
- Lesione intraossea - colpisce una o più articolazioni in cui si formano aree di tessuto fibroso, in rari casi il tessuto osseo degenera lungo tutta la sua lunghezza; Ma lo strato superiore (corticale) rimane invariato, quindi non c'è curvatura;
- Patologia completa (totale): la degenerazione colpisce tutte le parti dell'articolazione, compreso il canale midollare e lo strato superiore. Ciò porta alla deformazione degli arti e alle fratture da stress. Molto spesso, questa forma di displasia si sviluppa nelle ossa tubolari lunghe;
- Il tumore è una forma rara della malattia, caratterizzata da una forte crescita del tessuto fibroso;
- La sindrome di Albright è un'altra patologia correlata. Questa malattia comprende tre segni caratteristici: displasia fibrosa, iperpigmentazione focale (melanosi) del derma e pubertà precoce. Accompagnato da deformazioni irreversibili dello scheletro e degli arti, nonché da disfunzioni di organi e sistemi interni;
- La displasia fibrosa cartilaginea comporta la sostituzione del tessuto cartilagineo con tessuto fibroso. Può trasformarsi in condrosarcoma;
- La displasia fibrosa calcificante della tibia viene diagnosticata estremamente raramente ed è irreversibile.
Sintomi
Le deformità congenite visivamente evidenti sono rare. Di solito, i primi sintomi di displasia fibrosa dell'articolazione del ginocchio o di altre articolazioni compaiono durante l'infanzia sullo sfondo di melanosi cutanea, disturbi cardiovascolari e squilibrio ormonale.
I sintomi della malattia sono molto diversi. Ma nella maggior parte dei casi, i pazienti lamentano dolore e curvatura ossea. Spesso la malattia viene diagnosticata dopo una frattura abituale (patologica).
Più suscettibili alla displasia fibrosa:
- Ossa tubolari: femore, tibia, perone, omero, ulna e radio;
- Piatto: ossa del bacino, della colonna vertebrale, del cranio, delle costole e dei piedi.
Il livello di curvatura è determinato dalla localizzazione dei focolai fibrosi. Il danno alle ossa tubolari delle mani porta alla loro espansione; lo sviluppo della displasia delle dita è accompagnato dal loro accorciamento.
La displasia fibrosa delle articolazioni delle gambe porta alla loro deformazione, causata dal peso corporeo. Il femore nel 50% dei casi si accorcia e assume l'aspetto di un “boomerang” o di una “mazza da hockey”. Il grande trocantere del femore si solleva verso l'articolazione pelvica, provocando deformità e zoppia del collo del femore. L'altezza di un paziente con displasia può diminuire di 1-10 centimetri.
La displasia fibrosa del perone non porta alterazioni dell'arto, ma quando la lesione è nella tibia si registra una deformità a sciabola, un rallentamento della crescita ossea e un accorciamento dell'arto.
La patologia delle articolazioni sciatica e iliaca provoca la curvatura delle ossa pelviche, che provoca condizioni patologiche della colonna vertebrale - scoliosi o cifosi.
Interessante!
Con il danno simultaneo al femore e all'osso pelvico, il carico sulla colonna vertebrale aumenta a causa dello spostamento dell'asse corporeo.
Con la forma monoosa della displasia, i sintomi sono meno gravi. È possibile sviluppare dolore, zoppia e affaticamento quando si sottopone a stress l'articolazione danneggiata. Vengono diagnosticate anche fratture patologiche.
Diagnostica
La diagnosi di patologia include:
- Intervista al paziente;
- Esame iniziale;
- Esame radiografico.
Per la diagnosi sono importanti le fratture frequenti, dette patologiche, così come il dolore persistente e l'accorciamento della gamba.
Una nota!
La patologia non è sempre accompagnata dal dolore. È presente nel 50% degli adulti ed è completamente assente nei pazienti giovani. Ciò si spiega con la costante ristrutturazione delle ossa dei bambini man mano che crescono e con la loro elevata capacità compensatoria.
Utilizzando una radiografia è possibile confermare o smentire il sospetto di displasia. Le caratteristiche caratteristiche della displasia alla radiografia includono:
- Il tessuto osseo ricorda il “vetro smerigliato”;
- Si alternano fuochi luminosi e aree di compattazione;
- Macchie sull'osso;
- Evidente curvatura dell'articolazione interessata.
Dopo aver trovato un focolaio di tessuto fibroso, è necessario assicurarsi che non vi siano altre lesioni asintomatiche. Pertanto, viene eseguito un esame radiografico di tutte le ossa. E se necessario, viene eseguita una TAC.
Se i sintomi sono vaghi, la diagnosi finale viene fatta solo dopo l'osservazione a lungo termine della dinamica della patologia. È importante differenziare la malattia dalle patologie oncologiche e dalla tubercolosi ossea. A questo scopo prescrivono:
- Consultazione con un fisiatra;
- Test per la tubercolosi;
- Esame della ghiandola paratiroidea.
Terapia
Il trattamento conservativo di solito non è efficace. Pertanto, una volta chiarita la diagnosi, il paziente viene preparato per l’intervento chirurgico. Svolgere:
- Osteotomia;
- Resezione (escissione) della parte interessata dell'articolazione e sostituzione con un impianto;
- Allungamento osseo (se viene registrato l'accorciamento degli arti).
Quando si diagnostica la varietà poliostotica, la chirurgia non è indicata. Il trattamento per questo modulo include:
- Corso di massaggio speciale;
- Fisioterapia;
- Fisioterapia.
Previsioni e misure preventive
Le cause esatte della malattia non sono completamente comprese, quindi le misure preventive includono il rispetto delle regole generali:
- Rifiuto delle cattive abitudini;
- Mangiare sano;
- Visite sistematiche dal ginecologo durante la gravidanza;
- Controllo del peso;
- Evitare stress eccessivi sulle articolazioni;
- Esame preventivo sistematico da parte di un medico.
La prognosi per i pazienti con displasia fibrosa è positiva nella maggior parte dei casi. L'eccezione è la varietà poliostotica, che porta a deformità irreversibili. Lo sviluppo della patologia in un tumore benigno è osservato nel 4% e in una neoplasia maligna - solo nello 0,2% dei casi.
Displasia fibrosa.
Nel 1927 V.R. Al 19 ° Congresso dei chirurghi russi, Braitsev fu il primo a fornire in dettaglio una descrizione accurata del quadro clinico, radiologico e microscopico delle ossa modificate e riferì sulla struttura microscopica del focolaio della displasia fibrosa. Credeva che la base della malattia fosse "la deviazione delle funzioni del mesenchima osteoblastico... Il mesenchima osteoblastico crea osso con una struttura incompleta". Pertanto, dobbiamo essere d'accordo con l'opinione di T.P. Vinogradova (1973), che ci sono molte più ragioni per assegnare il nome V.R. alla displasia ossea fibrosa. Braitsev, anziché chiamarla Lichtenstein o malattia di Lichtenstein-Jaffe, che ha solo chiarito e sviluppato ulteriormente le disposizioni della V.R. Braitseva.
VR Braitsev descrisse la displasia fibrosa anche nelle riviste “New Surgery” (1928) e “Archiv klinische Chirurgi” (1928), cioè 10 anni prima di J. Lichtenstein, che nel 1938 riferì di questa malattia e nel 1942 descrisse 15 delle sue osservazioni.
È necessario correggere questa ingiustizia storica: il merito di V.R. La scoperta da parte di Braitseva di una nuova entità nosologica - la displasia ossea fibrosa - è ovvia.
VR Braitsev nel 1927 al XIX Congresso dei chirurghi russi fece anche un rapporto sull'osteodistrofia locale - osteodistrofia fibrosa localisata (cistica). Ha detto: "I chirurghi sono particolarmente interessati al lato pratico della questione, ma l'opportunità delle misure chirurgiche per l'osteodistrofia locale può basarsi solo sulla completa chiarezza dell'essenza della malattia". Conoscendo bene la letteratura mondiale su questo tema, sulla base di tre proprie osservazioni, propone una nuova teoria originale sull'origine e l'essenza della malattia, la identifica come un'unità nosologica indipendente, dimostra le ragioni dello sviluppo del tessuto fibroso cisti e raccomanda metodi di trattamento. La descrizione del quadro morfologico della displasia fibrosa è insolitamente accurata. In conclusione, trae le seguenti conclusioni.
1. L'essenza dell'osteodistrofia fibrosa è la deviazione funzionale del mesenchima osteoblastico durante lo sviluppo osseo nel periodo embrionale, a seguito della quale, fin dall'inizio, si crea un osso peculiare con midollo osseo fibroso, capace di crescere e produrre “osteoide tessuti e ossa di tipo incompleto”.
2. Tale deviazione nella funzione del mesenchima osteoblastico può verificarsi in aree isolate di un singolo osso o può diffondersi all'intero osso e persino a molte ossa dello scheletro.
3. La crescita del tessuto fibroso è attiva, ma l'energia di crescita è diversa nei diversi casi. In alcuni casi, il processo procede silenziosamente, lentamente, in altri - rapidamente, accompagnato da un ampio polimorfismo cellulare, che lo avvicina morfologicamente al sarcomatoso.
4. Le cisti ossee solitarie, secondo i dati ottenuti da molti autori, si sviluppano a causa dell'osteodistrofia fibrosa dovuta all'edema e alla liquefazione delle escrescenze fibrose centrali, e anche, possibilmente, a causa di emorragie nel tessuto fibroso.
V. R. Braitsev consiglia di eseguire una resezione sottoperiostale longitudinale con sostituzione del difetto con autoinnesti, perché "il tessuto fibroso patologico, come si può vedere dall'esame istologico, penetra nel guscio osseo fino al periostio".
Chirurghi eminenti come I.I. Grekov, S.P. Fedorov, N.N. Petrov, ma dai loro discorsi è chiaro che hanno sottovalutato i dati unici ottenuti da V.R. Braitsev, - la sua scoperta di una nuova unità nosologica. Tutti questi chirurghi, come N.N. Terebinsky e T.N. Krasnobaev, parlavano solo delle cisti ossee, che spesso incontravano e per le quali a volte eseguivano operazioni.
Sotto la nostra osservazione in ospedale c'erano 245 pazienti con displasia fibrosa; il numero di pazienti con lesioni poliostotiche era significativamente maggiore del numero di pazienti con processo monostotico che necessitavano di trattamento chirurgico.
Secondo la letteratura, le forme monoostotiche e poliostotiche di displasia fibrosa si osservano quasi altrettanto spesso, tuttavia, secondo M.K. Klimova (1970), la forma poliostotica è ancora un po' più comune, e anche M.V. Volkov (1968, 1985).
Clinica. I pazienti nascono raramente con gravi deformità scheletriche. I sintomi della displasia fibrosa compaiono solitamente durante l'infanzia e sono caratterizzati da diversità: si tratta di un dolore lieve, solitamente ai fianchi, o della comparsa di deformità e del suo aumento, o di una frattura patologica dovuta a un trauma grave e inadeguato, e la diagnosi corretta è non sempre realizzato.
Nella forma poliostotica della displasia fibrosa, sono più spesso colpiti la tibia, il femore, il perone, l'omero, il radio e l'ulna. La frequenza del danno (in ordine decrescente) delle ossa piatte: ossa pelviche, ossa del cranio, vertebre, costole, scapola. Le ossa del piede e della mano (ma non le ossa del polso) sono relativamente spesso colpite.
I bambini affetti dalla sindrome di Albright talvolta nascono con gravi deformità e, ovviamente, con una tipica macchia pigmentata. Dopo la prima manifestazione della malattia, sia i segni clinici che quelli radiologici possono progredire e la forma intraossea della malattia può poi svilupparsi in una forma che colpisce l'intero strato corticale o nell'area di uno dei focolai, più spesso nella parte superiore fine del femore o lungo tutta la diafisi, che indica varie attività del processo displastico. Le epifisi delle ossa, di regola, non sono interessate. La progressione del processo nei bambini e nei giovani è spesso accompagnata da fratture. Secondo le osservazioni di A.I. Snetkova (1984), in un paziente operato a 4 anni (resezione marginale, rimozione di tessuto fibroso, alloplastica ossea), dopo 7 anni si è osservata una ristrutturazione degli alloinnesti con zone di loro lisi, causata dalla crescita di nuovi focolai patologici. Pertanto, esiste uno schema programmato nello sviluppo della displasia: in alcuni pazienti si sviluppa tessuto fibroso displastico in aree dell'osso che precedentemente apparivano normali radiograficamente.
L.N. Furtseva et al. (1991) hanno rivelato disturbi significativi nella funzione glucocorticoide della corteccia surrenale in pazienti con displasia fibrosa: “Il livello di calcio in tutti i tipi di malattia è aumentato, ma non in proporzione all'entità del danno al tessuto osseo; allo stesso tempo, l'escrezione di calcio nelle urine è ridotta rispetto alla norma. La diminuzione è più pronunciata nella forma poliostotica che in quella monoostotica. Nelle forme limitate della malattia, la fosfaturia è ridotta nelle lesioni estese del tessuto osseo, si osserva solo una tendenza al ribasso; L'azoto amminico totale e l'idrossiprolina totale nelle urine aumentano nei processi estesi, e nella sindrome di Albright e nella forma poliostotica con lesioni diffuse, l'escrezione di aminoacidi è significativamente più elevata.
Sistema cardiovascolare: più spesso nei pazienti con forma poliostotica si osserva tachicardia sinusale - 96-140 al minuto, meno spesso si nota aritmia sinusale sull'ECG e nella maggior parte dei pazienti l'ipotensione arteriosa è 115/60 e persino 95/50 mm Hg, con In alcuni pazienti, il metabolismo nel muscolo cardiaco viene interrotto. Si nota un aumento della VES: nei pazienti con forma monoostotica - fino a 15-27 mm/h, con forma poliostotica - fino a 22-45 mm/h. Durante lo studio della funzione surrenale, è stato determinato il contenuto di 11-idrossicorticosteroidi (11-OX) nel plasma; Nei pazienti con forma poliostotica è stato rivelato un disturbo dello stato funzionale della corteccia surrenale, come evidenziato da un aumento meno pronunciato del livello di 11-OX totale e attivo nel plasma sanguigno e da una risposta indebolita o paradossale alla somministrazione dell’ormone adrenocorticotropo (ACTH).
Un paziente di 30 anni affetto dalla forma poliostotica morì durante un intervento chirurgico nel 1974 a causa di un improvviso calo della pressione sanguigna; il secondo, all'età di 19 anni, morì dopo un intervento chirurgico nel 1978 per insufficienza cardiaca polmonare progressiva. Durante l'esame patologico, oltre ai tipici cambiamenti nelle ossa, sono stati rilevati cambiamenti simili: ovaie policistiche, adenomatosi della corteccia surrenale, ghiandola tiroidea, ghiandola pituitaria anteriore e iperplasia del timo.
A.I. Morozov, V.P. Ivannikov (1972), studiando un caso di “malattia di Albright” in un paziente di 16 anni, identificò cambiamenti nelle strutture cerebrali profonde della linea mediana sull’elettroencefalogramma. A.G. Povarinsky, Z.K. Bystrov, con l'aiuto di studi encefalografici condotti parallelamente a studi neurologici, ha stabilito disfunzioni profonde e caratteristiche del cervello e l'instabilità dell'omeostasi cerebrale come risultato di un disturbo dei meccanismi regolatori situati nell'area delle strutture profonde. Tutto ciò ci ha costretto negli ultimi 20 anni ad esaminare attentamente i pazienti con displasia fibrosa, soprattutto con la forma poliostotica.
In un certo numero di pazienti, questa displasia si manifesta in modo latente. Abbiamo consultato un professore-chirurgo di 62 anni, nel quale per la prima volta è stata scoperta casualmente sulle radiografie la displasia fibrosa dell'intero femore sinistro e della tibia, e non abbiamo stabilito alcuna indicazione per il trattamento chirurgico. Non crediamo che tutti i pazienti affetti da displasia fibrosa debbano essere operati, soprattutto quelli con forma intraossea. Le indicazioni per l'intervento chirurgico sono il dolore causato da deformazione progressiva, fratture da stress, presenza di cisti con un forte assottigliamento dello strato corticale, zoppia, accorciamento dell'arto, compressione del midollo spinale, ecc. Se c'è una frattura nella sede della cisti, eseguiamo un trattamento conservativo, aspettiamo 8 mesi dopo la guarigione e se la cisti rimane della stessa dimensione o progredisce, operiamo.
M.V. Volkov (1985) distingue tra forme poliostotiche, monoossee e regionali di displasia fibrosa e in base alla natura dei cambiamenti nell'osso - focali e diffusi. Offriamo una classificazione clinica con una descrizione più dettagliata delle caratteristiche di ciascuna forma, utilizzando il concetto da noi proposto di "memoria della forma ossea", tuttavia, va tenuto presente che la displasia fibrosa è un processo patologico che presenta innumerevoli varianti e forme transitorie .
Classificazione clinica della displasia ossea fibrosa (secondo S.T. Zatsepin)
La classificazione proposta include le seguenti forme. I. Forma intraossea di displasia fibrosa: i focolai di tessuto fibroso possono essere singoli, multipli, occupare qualsiasi parte dell'osso o l'intero osso, tuttavia, lo strato corticale può essere assottigliato, ma mantiene una struttura normale - la forma delle ossa rimane corretto, poiché non vi è alcuna compromissione della memoria nelle forme ossee. Possono essere colpiti uno o più ossa di diversi segmenti dell'arto, ad es. il processo è monoostotico o poliostotico.
Un intervento adeguato è la rimozione completa del tessuto fibroso mediante resezione marginale dell'osso della lunghezza richiesta, rimozione del tessuto fibroso e sostituzione della cavità con omoinnesti ossei conservati. L'intervento può essere considerato radicale se le pareti della cavità sono intatte il tessuto fibroso individuato viene accuratamente lavorato con uno scalpello. In questa forma, le cisti si sviluppano spesso al centro di masse fibrose; le cisti raggiungono la corticale assottigliandola, dando spesso luogo a fratture patologiche (Fig. 15.1). Più spesso ricorrono a tattiche in due fasi: 1) viene applicata la trazione scheletrica - i frammenti guariscono bene, poiché lo strato corticale e il periostio sono normali; spesso le cavità cistiche scompaiono e, in caso contrario, 2) eseguire la resezione marginale, la rimozione delle masse fibrose, l'alloplastica ossea o eseguire un'operazione secondo la nostra tecnica (vedi sotto).
II. Il processo patologico coinvolge tutti gli elementi dell'osso: l'area del canale midollare, lo strato corticale, la spongiosi delle metafisi, le ossa lunghe sono spesso colpite per tutta la lunghezza, ma la gravità del processo patologico varia; di solito è una lesione poliostotica. Il danneggiamento di tutti gli elementi che compongono l'osso (danno totale) riduce la sua resistenza meccanica, portando a deformazioni graduali e fratture per fatica. Con questa forma di displasia fibrosa SI ESPRESSA LA SINDROME DELLA MEMORIA OSSEA. Non esistono interventi chirurgici radicali adeguati (ad eccezione della rimozione dell'intero osso interessato. Sono ampiamente utilizzate osteotomie correttive ortopediche con alloplastica e strutture metalliche.
Crescite simili a tumori di focolai di tessuto fibroso possono raggiungere grandi dimensioni, il che indica una maggiore massa del processo, ma sono raramente osservate.
III. Forme tumorali di displasia fibrosa.
IV. La sindrome di Albright è una forma speciale di displasia, quando, insieme alla forma poliostotica o quasi generalizzata - displasia ossea fibrosa totale - si osservano numerosi disturbi endocrini con pubertà precoce nelle donne, campi di pigmentazione sulla pelle, proporzioni corporee alterate e spesso bassa statura; gravi deformazioni delle ossa degli arti, del bacino, della colonna vertebrale, delle ossa della base del cranio, cambiamenti pronunciati nel sistema cardiovascolare e in altri sistemi corporei. Durante la vita, il processo progredisce, le deformazioni ossee aumentano gradualmente. La sindrome della memoria della forma ossea compromessa è pronunciata.
Esistono molte varianti di questa forma della malattia; nessun paziente ne ripete un'altra, ma è necessariamente diversa in qualche modo. Lo studio della struttura morfologica del tessuto fibroso e osseo patologico in pazienti con questa forma di displasia ha mostrato che le varie manifestazioni cliniche di questa displasia corrispondono a un'ampia varietà di modelli istologici, quindi il compito dei ricercatori è quello di studiare e confrontare clinico-radiomorfologico , che consentirà senza dubbio di identificare sottogruppi di pazienti.
V. La displasia ossea fibroso-cartilaginea come forma speciale nel nostro paese è stata isolata e descritta da M.A. Berglezov e N.G. Shulyakovskaya nel 1963, che osservò un paziente con un quadro morfologico clinico e radiografico pronunciato. Nelle osservazioni descritte, le manifestazioni di displasia cartilaginea vengono alla ribalta, i casi di sviluppo del condrosarcoma non sono rari;
VI. Il fibroma calcificante delle ossa lunghe appartiene a un tipo speciale di displasia fibrosa, descritta nel 1958 da N.E. Schlitter, R.L. Kempsom (1966), che lo studiò al microscopio elettronico.
QUELLO. Goerghen et al. (1977) descrissero 2 pazienti, uno dei quali ebbe una recidiva dopo il curettage e fu resecato insieme al periostio per 10 cm, ma dopo 1 anno fu notata una piccola recidiva all'estremità della parte inferiore della tibia - questo è stata l'unica osservazione in caso di recidiva del tumore. Sono stati descritti un totale di 8 tumori di questo tipo, localizzati nella tibia e un tumore nell'omero. La struttura istologica dei tumori è identica a quella osservata nelle ossa del cranio facciale e delle mascelle.
S.T.Zatsepin
Patologia ossea dell'adulto
R. M. Rzaev
La displasia fibrosa è una malattia ossea, che si basa su un processo simile a un tumore associato allo sviluppo improprio del mesenchima osteogenico. In questo caso, si verifica il processo di sostituzione del tessuto osseo con tessuto fibroso, a seguito del quale si verifica la deformazione ossea. Le ragioni per lo sviluppo di questa patologia non sono sufficientemente chiare. A seconda della distribuzione della lesione, si distinguono forme monoostotiche (quando un osso è coinvolto nel processo) e poliostotiche (quando sono interessate più ossa) di displasia fibrosa.
Nella letteratura disponibile non siamo riusciti a trovare informazioni sulla displasia fibrosa della base cranica e pertanto riteniamo opportuno presentare la nostra osservazione di un paziente con displasia fibrosa poliostotica della base cranica, sul quale abbiamo eseguito un intervento utilizzando il nostro propria tecnica per rimuovere la lesione.
Il paziente N., 35 anni, fu ricoverato nel reparto il 14 marzo 1991 lamentando mal di testa costante, protrusione del bulbo oculare sinistro e visione doppia. La malattia iniziò all'inizio del 1989, quando il paziente notò una protrusione del bulbo oculare sinistro. Successivamente questo fu accompagnato da mal di testa e visione doppia. Durante la visita dal medico, alla paziente è stato diagnosticato un glaucoma e le è stato prescritto un trattamento sintomatico, che non ha portato ad un miglioramento delle sue condizioni. Una TAC ha rivelato un tumore alla base del cranio. Il paziente è stato indirizzato al nostro reparto per decidere sulla possibilità di un intervento chirurgico.
Al momento del ricovero le condizioni del paziente erano soddisfacenti; Non sono stati riscontrati cambiamenti patologici negli organi interni e ENT. I risultati degli esami del sangue e delle urine erano senza alterazioni patologiche.
Conclusione dell'oculista: esoftalmo sinistro, sintomi iniziali di ristagno del capezzolo del nervo ottico. La vista è soddisfacente (Vis.OD = 1,0; OS = 0,9). La mobilità dei bulbi oculari non è limitata.
Le tomografie computerizzate mostrano una compattazione compatta, caratteristica della malattia ossea displastica, localizzata principalmente nel corpo dell'osso sfenoide a sinistra (Fig. 1, a. Sono visibili foci di “gonfiore” dell'osso sfenoide, a causa dei quali il suo corpo acquisisce una forma arrotondata. La lesione copre la superficie superiore, anteriore e laterale della superficie maggiore e inferiore dell'ala minore dell'osso sfenoide orbita sinistra, si notano la fessura orbitaria superiore ristretta, la compressione del nervo ottico e lo spostamento anteriore del bulbo oculare sinistro (Fig. 1, b).
Il 25 aprile 1991 è stata eseguita un'operazione per rimuovere un focolaio di displasia fibrosa della base cranica utilizzando un approccio attraverso la fossa infratemporale a sinistra. In anestesia endotracheale è stata praticata un'incisione cutanea arcuata, partendo dal bordo anteriore della regione temporale sinistra e terminando a livello del padiglione auricolare. Dopo la dissezione dello strato di grasso sottocutaneo, il lembo cutaneo è stato separato e spostato fino all'esposizione della regione temporo-parotidea. Successivamente, l'arco zigomatico su entrambi i lati viene sezionato obliquamente, dopo di che il muscolo temporale viene separato dall'osso (in seguito il muscolo viene utilizzato per obliterare la cavità postoperatoria). Dopo lo spostamento verso il basso del muscolo temporale mobilizzato e dell'arco zigomatico reciso (insieme al muscolo massetere attaccato), sono stati sezionati i legamenti dell'articolazione temporo-mandibolare. Quindi, utilizzando un divaricatore di Gosse inserito nella ferita chirurgica (un gancio del divaricatore viene inserito nella fossa articolare), la testa della mandibola viene spostata verso il basso. Ciò ha permesso di espandere la ferita chirurgica, separare i tessuti molli dalla base del cranio ed esporre la fossa infratemporale. L'arteria media isolata della dura madre viene coagulata e sezionata vicino al forame spinoso. Partendo dalla superficie laterale della grande ala dello sfenoide, sotto il controllo del microscopio operatorio, mediante fresa ed elettroaspirazione, sono stati asportati la cresta infratemporale e il bordo zigomatico della superficie temporale della grande ala dello sfenoide . Le ossa furono resecate fino a esporre la superficie della dura madre del lobo temporale del cervello. In questa fase dell'intervento è stata raggiunta la fossa infratemporale, sulla cui parete superiore (a livello del corpo dell'osso sfenoide) era presente un focolaio grigio-biancastro di tessuto patologico. La parte visibile del tessuto patologico raggiungeva la parte anteriore del fondo della fossa temporale (la superficie laterale della grande ala dell'osso sfenoide). Utilizzando una fresa e cucchiai chirurgici (sotto il controllo di un microscopio operatorio), sono stati rimossi i focolai di tessuto patologico insieme alle aree interessate della base del cranio e delle ossa dello scheletro facciale. È stato stabilito che le superfici superiore, anteriore e laterale della grande ala, così come la superficie inferiore della piccola ala dell'osso sfenoide, erano coinvolte nel processo patologico. Dopo l'emostasi, la cavità postoperatoria è stata cancellata dal muscolo temporale, che ha anche riempito i difetti sorti dopo l'intervento chirurgico sulla base del cranio, le parti posteriori delle pareti superiore ed esterna dell'orbita. Quindi la testa della mascella inferiore viene riportata nella fossa articolare e l'arco zigomatico viene fissato con un filo nella sua posizione originale. Nella fase finale dell'operazione, la ferita chirurgica è stata drenata e sono state posizionate delle suture sulla pelle.
L'esame istopatologico ha rivelato tessuto connettivo fibroso che sostituisce il midollo osseo nel campione. In alcune sue aree si individuano fasci ossei scarsamente calcificati, che formano osso spugnoso di varia maturità. In alcuni punti, il tessuto fibroso è costituito da fasci di fibre di collagene mature e cellule del fuso posizionate in modo caotico. Conclusione: displasia fibrosa.
Il paziente è stato dimesso dall'ospedale in buone condizioni il 31° giorno dopo l'intervento. Esoftalmo e diplopia sono scomparsi. Quando esaminato 6 e 12 mesi dopo l'intervento chirurgico, il paziente non ha presentato lamentele. Le tomografie computerizzate non hanno mostrato segni di recidiva della malattia (Fig. 2). L'unico difetto evidente nel paziente dopo l'intervento era una leggera deformazione (sotto forma di depressione) e una cicatrice poco appariscente nella regione temporale, nascosta sotto i capelli (Fig. 3).
Pertanto, questa osservazione indica la possibilità di sviluppare una displasia fibrosa poliostotica alla base del cranio, il cui riconoscimento è difficile. L'intervento chirurgico è il metodo di trattamento ottimale per i pazienti con questa patologia. L'intervento radicale può essere ottenuto mediante intervento chirurgico (secondo l'autore) utilizzando un approccio attraverso la fossa infratemporale. La TC consente di identificare il tessuto patologico in una determinata patologia, determinare i confini della diffusione della lesione e giudicare l'efficacia del trattamento.
Letteratura
1. Rzaev R.M. Un metodo per rimuovere un tumore alla base del cranio. Brevetto 980024 del 9 novembre 1992. Bollettino ufficiale del Comitato brevetti e licenze per la scienza e la tecnologia della Repubblica dell'Azerbaigian. Baku 1998; 37-38.
2. Strukov A.I., Serov V.V. Anatomia patologica: libro di testo. Ed.
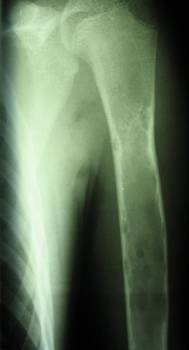
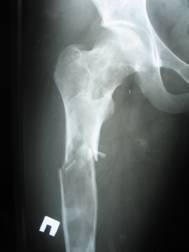 Displasia fibrosa- una malattia caratterizzata da un disturbo dello sviluppo (displasia) dello scheletro, in cui l'osso normale viene sostituito da tessuto fibroso con elementi di osso displastico. Esistono forme monoostotiche (circa l'85% dei casi), monomeliche (sono interessate diverse ossa adiacenti di un arto, spalla o cintura pelvica) e poliostotiche (circa il 5% dei casi). La displasia fibrosa come anomalia dello sviluppo è talvolta combinata con disturbi extrascheletrici: 1) la forma poliostotica della lesione è combinata con pubertà prematura e macchie di pigmento sulla pelle sotto forma di macchie di caffè (sindrome di Albright); 2) una combinazione di displasia fibrosa con mixomi dei tessuti molli (sindrome di Mazabraud). Si manifesta a qualsiasi età, più spesso nelle prime 3 decadi di vita (70% dei pazienti). La forma monoossea è un po' più comune nei maschi; con la forma poliostotica, il rapporto tra maschi e femmine è 2:1. Qualsiasi osso è interessato, la sede più comune: la parte prossimale del femore, tibia e omero, costole, cranio. ossa, colonna cervicale. Le ossa della colonna vertebrale sono colpite solo nel 2,5% dei casi.
Displasia fibrosa- una malattia caratterizzata da un disturbo dello sviluppo (displasia) dello scheletro, in cui l'osso normale viene sostituito da tessuto fibroso con elementi di osso displastico. Esistono forme monoostotiche (circa l'85% dei casi), monomeliche (sono interessate diverse ossa adiacenti di un arto, spalla o cintura pelvica) e poliostotiche (circa il 5% dei casi). La displasia fibrosa come anomalia dello sviluppo è talvolta combinata con disturbi extrascheletrici: 1) la forma poliostotica della lesione è combinata con pubertà prematura e macchie di pigmento sulla pelle sotto forma di macchie di caffè (sindrome di Albright); 2) una combinazione di displasia fibrosa con mixomi dei tessuti molli (sindrome di Mazabraud). Si manifesta a qualsiasi età, più spesso nelle prime 3 decadi di vita (70% dei pazienti). La forma monoossea è un po' più comune nei maschi; con la forma poliostotica, il rapporto tra maschi e femmine è 2:1. Qualsiasi osso è interessato, la sede più comune: la parte prossimale del femore, tibia e omero, costole, cranio. ossa, colonna cervicale. Le ossa della colonna vertebrale sono colpite solo nel 2,5% dei casi.

 Quadro clinico. Dolore di varia intensità, curvatura e deformazione delle ossa, fratture patologiche.
Quadro clinico. Dolore di varia intensità, curvatura e deformazione delle ossa, fratture patologiche.
Radiografia - lesione litica con contorni chiari e bordi sclerotici; potrebbe esserci gonfiore dell'osso, una frattura patologica.
Patomorfologia.
Macroscopicamente: tessuto bianco-grigio denso con piccole inclusioni di densità ossea; il tessuto patologico è quasi sempre localizzato solo all'interno del canale midollare.
 Microscopia. Si formano stroma del tessuto connettivo con trabecole ossee di struttura primitiva, cambiamenti mixoidi, talvolta strutture di tipo cemento, focolai di tessuto condroide e cisti. Le trabecole ossee sono limitate solo occasionalmente dagli osteoblasti. Atipie e mitosi citologiche non sono tipiche. In casi molto rari si può osservare atipia cellulare associata ad alterazioni degenerative o regressive.
Microscopia. Si formano stroma del tessuto connettivo con trabecole ossee di struttura primitiva, cambiamenti mixoidi, talvolta strutture di tipo cemento, focolai di tessuto condroide e cisti. Le trabecole ossee sono limitate solo occasionalmente dagli osteoblasti. Atipie e mitosi citologiche non sono tipiche. In casi molto rari si può osservare atipia cellulare associata ad alterazioni degenerative o regressive.
Diagnosi differenziale. Displasia osteofibrosa, granuloma riparativo a cellule giganti dell'osso, morbo di Paget, osteosarcoma centrale ben differenziato, osteosarcoma parostale.
Trattamento. Resezione marginale dell'osso seguita da chirurgia plastica del difetto. In caso di gravi deformità ossee, la resezione marginale dovrebbe essere combinata con osteotomie correttive con osteosintesi extraossea o intraossea.
Esodo favorevole; la malignità della displasia fibrosa è osservata nello 0,4% dei casi, con la sindrome di Albright - nel 4% dei casi, più spesso all'età di 30-40 anni. Nella maggior parte dei casi, la displasia fibrosa è maligna, localizzata nelle ossa dello scheletro facciale (mascella inferiore e superiore), quindi nel femore e nelle ossa pelviche.
È un'anomalia non ereditaria dello sviluppo osseo in cui il normale tessuto osseo viene sostituito da tessuto fibroosseo. Questa condizione fu descritta per la prima volta nel 1942 da Lichtenstein e Jaffe. Di conseguenza, la displasia fibrosa è talvolta chiamata malattia di Lichtenstein-Jaffe. Il processo patologico può essere localizzato in una o più ossa.
La displasia fibrosa localizzata può verificarsi come parte della sindrome di McCune-Albright o di Mazabraud. La displasia fibrosa può anche svilupparsi in connessione con altre disfunzioni endocrine, ad esempio con ipertiroidismo, iperparatiroidismo, acromegalia, diabete mellito, sindrome di Cushing.
Displasia fibrosa. Epidemiologia
La displasia fibrosa rappresenta circa il 5% di tutte le lesioni ossee benigne. Tuttavia, la reale incidenza non è nota poiché molti pazienti sono asintomatici. La displasia fibrosa localizzata rappresenta il 75-80% dei casi.
La displasia fibrosa è una lesione a crescita lenta che di solito appare durante i periodi di crescita ossea e quindi può essere spesso osservata in soggetti nella prima e nella tarda adolescenza. La displasia fibrosa, che si sviluppa in diverse ossa, rappresenta il 20-25% di tutti i casi e i pazienti affetti da questa forma tendono a manifestare le manifestazioni della malattia in età leggermente precoce (età media 8 anni).
La gravidanza può causare un aumento della crescita della lesione. Maschi e femmine sviluppano la malattia con la stessa frequenza, sebbene la variante multipla, associata alla sindrome di McCune-Albright, sia più comune nelle donne.
Displasia fibrosa. Cause
La displasia fibrosa è causata da una mutazione somatica nel gene GNAS1, situato sul cromosoma 20q13.2-13.3. Questo gene codifica per la subunità alfa della proteina stimolatoria G, Gsα. Come risultato di questa mutazione, l’aminoacido arginina (nella proteina) in posizione 201 (R201) viene sostituito dall’aminoacido cisteina (R201C) o dall’istidina (R201H).
Questa proteina anomala stimola l'adenosina monofosfato ciclico (AMP) G1 e gli osteoblasti (cellule) a sintetizzare il DNA a una velocità maggiore rispetto alle cellule normali. Ciò si traduce nella formazione di una matrice ossea fibrosa e disorganizzata, che produce tessuto osseo primitivo che non è in grado di maturare in osso lamellare. Anche il processo di mineralizzazione stesso è anormale.
Displasia fibrosa. Foto
Displasia fibrosa. Sintomi e manifestazioni
I pazienti con lesioni piccole e solitarie possono non presentare alcun sintomo o segno e le loro anomalie ossee possono essere scoperte incidentalmente durante gli esami radiografici per ragioni completamente diverse. Tuttavia, il dolore osseo, il gonfiore e la dolorabilità sono i sintomi e le manifestazioni più comuni nei pazienti sintomatici.
Anomalie endocrine possono essere la manifestazione iniziale in alcuni pazienti.
Le sedi scheletriche più comuni in cui si sviluppano displasie fibrose solitarie comprendono le costole, il femore prossimale, le ossa craniofacciali e tipicamente la mascella posteriore. Queste singole lesioni di solito coinvolgono solo un piccolo segmento dell’osso, ma in alcuni casi questo tessuto fibroso può occupare l’intera lunghezza dell’osso.
Nella displasia fibrosa multipla, la malattia può svilupparsi da almeno due ossa fino a oltre il 75% delle ossa dello scheletro. Questa forma di displasia fibrosa si verifica più spesso nelle ossa della coscia, della parte inferiore della gamba, del bacino e delle gambe. Altre sedi in cui si sviluppa questa malattia, ma meno frequentemente, includono: le costole, il cranio e le ossa degli arti superiori. E ancora più raramente (e anche insolitamente) questa forma della malattia può svilupparsi nella zona lombare, cervicale e delle clavicole.
Le deformità fisiche più comuni di questa malattia sono le discrepanze nella lunghezza delle gambe, l'asimmetria facciale e le deformità delle costole. La frattura è la complicanza più comune della displasia fibrosa di tutti e due i tipi. Le fratture ossee si verificano in più della metà dei pazienti. Molti individui sviluppano anche deformità delle ossa portanti. Quasi il 75% dei pazienti affetti da displasia fibrosa soffre di dolore, deformità ossee o fratture patologiche.
Trasformazione maligna
La trasformazione maligna della displasia fibrosa è molto rara, con un'incidenza riportata di circa lo 0,4-4%. Più della metà dei pazienti con trasformazione maligna hanno ricevuto radioterapia.
I tumori maligni più comuni sono gli osteosarcomi, i fibrosarcomi e i condrosarcomi. La maggior parte dei pazienti ha > 30 anni. La maggior parte di questi tumori cancerosi si sviluppano nelle ossa del cranio, seguite da anche, gambe e bacino. Il tasso di trasformazione maligna è più elevato nelle lesioni singole che in quelle multiple.
Displasia fibrosa. Diagnostica
Radiografia convenzionale
Durante l'esecuzione della radiografia, gli operatori della macchina e altri specialisti competenti in materia possono rilevare la displasia fibrosa come una lesione intramidollare e ben definita nell'area della diafisi o della metafisi dell'osso. Le lesioni possono variare da completamente lucenti a completamente sclerotiche. Tuttavia, la maggior parte delle lesioni presenta un caratteristico aspetto nebuloso o di vetro smerigliato. Il grado di opacizzazione su una radiografia è direttamente correlato all'istopatologia sottostante. Le lesioni più radiolucenti sono composte prevalentemente da elementi fibrosi, mentre le lesioni più radiopache contengono una percentuale maggiore di tessuto osseo. Inoltre, le lesioni possono essere circondate da uno spesso strato di tessuto reattivo sclerotico chiamato “pelle”.
Scintigrafia, TC e RM
La scintigrafia può essere utilizzata per determinare l’entità della malattia. Le lesioni fibrose attive, soprattutto nei pazienti giovani, aumentano significativamente l'assorbimento degli isotopi; tale assorbimento diventerà meno intenso man mano che la lesione matura;
Ma soprattutto, l'entità del danno viene dimostrata tramite una tomografia computerizzata. Alla TC, nella maggior parte dei pazienti la displasia fibrosa può essere facilmente distinta dalle altre lesioni nella diagnosi differenziale.
Displasia fibrosa. Trattamento
Le lesioni fibrose asintomatiche e radiologicamente caratteristiche richiedono solo l'osservazione clinica. Le radiografie devono essere eseguite ogni 6 mesi. Se nel paziente vengono rilevate nuove lesioni, i medici dovranno escludere la diagnosi di una forma multipla di questa malattia. Altrimenti, il paziente deve essere indirizzato a un endocrinologo per la diagnosi precoce di possibili disturbi sistemici.
I bifosfonati, in particolare il pamidronato, possono essere utilizzati per ridurre il dolore osseo nei pazienti sintomatici con displasia fibrosa solitaria. Una biopsia aperta può essere indicata per quei pazienti che presenteranno segni insoliti. La maggior parte dei pazienti necessiterà di un intervento chirurgico per correggere deformità, prevenire fratture patologiche o rimuovere lesioni sintomatiche.
Il trattamento per la trasformazione maligna dovrebbe basarsi sul sottotipo di sarcoma, ma la prognosi è generalmente sfavorevole.
Displasia fibrosa. Previsione
Il tasso di recidiva della displasia fibrosa, anche dopo curettage e innesto osseo, è elevato. Tuttavia, la maggior parte delle lesioni solitarie si stabilizza con la maturità scheletrica. Di norma, una forma singola non si trasforma in una forma multipla.
Le forme multiple possono essere molto gravi (nelle loro manifestazioni), ma si stabilizzano anche (molto spesso) durante la pubertà. Tuttavia, le deformità esistenti possono progredire.



